
בשנה שעברה התחלתי בסיקור רפואת העתיד עם הסבר מורחב בכל מה שקשור בטיפול הגנטי בכתבה "טיפול גנטי, צעדים ראשונים בעולם הרפואה העתידית" (לינק) ומאז ראינו במהלך שנת 2018 התפתחות רבה והצלחות בתחום מעניין זה.
בשנים האחרונות הפך בית חולים Nationwide Children’s Hospital, למרכז המחקר המתקדם בעולם לטיפול גנטי, וחממה לשיתופי פעולה עם חברות הביומד לקידום הטיפול הגנטי לניסויים קליניים מתקדמים. ההצלחה הראשונה הייתה בשיתוף הפעולה עם חברת Avexis לטיפול בתינוקות עם מחלת ה SMA1, אותה חברה אשר נרכשה בחודש אפריל 2018 ע"י נוברטיס תמורת 8.7 מליארד דולר. אבל שיתופי הפעולה המשמעותיים ביותר נחתמו בעיקר עם חברת Sarepta Therapeutic לקידום התוכנית לטיפול גנטי בחולי ה DMD והמשיכו בתוכנית השניה לטיפול הגנטי בחולי ה LGMD, והדרך קדימה נראית ורודה מתמיד.
בכתבה זו, מביא לכם את החידושים האחרונים בעולם הטיפול הגנטי אשר פורסמו ע"י מרכז המחקר של Nationwide Children’s Hospital, והביאו לקפיצה ענקית בטיפול הגנטי בבני אדם אשר מתחילים להראות איתותי הצלחה בניסויים הקליניים המתקדמים. כפי שכתבה Abbie Ruth ב pediatricsnationwide.org
שינוי תכונות הוירוס – צעד חשוב לעזרת המדע
רתימת יכולתו של הוירוס להעביר חומר גנטי לתא לטיפול, ריפוי או מניעה של מחלות היתה המטרה ארוכת הטווח של חוקרים העובדים בטיפול גנטי. במהלך 30 השנים האחרונות, המסע לריפוי גנטי היה טומן בחובו אתגרים ומחסומים רבים. אבל בשנת 2017, לאחר אינספור התחלות ועצירות לאורך הדרך, התחום חווה פריצות דרך רבות.
שום פריצת דרך לא זכתה לתשומת לב רבה יותר מאשר ההצלחה בתחום הטיפול הגנטי כאשר חוקרים מ Nationwide Children’s Hospital ואוניברסיטת אוהיו פרסמו את התוצאות המדהימות של הניסויים הקליניים שלב 1 של טיפול גנטי בסוג 1 (SMA1- Spinal Muscular Atrophy type 1) של ניוון שרירי השדרה בכתב העת המדעי- New England Journal of Medicine.
מחקר ה- SMA1 בשלב מוקדם, בהנהגתו של דר' ג'רי מנדל, החוקר הראשי של המרכז לטיפול גנטי במכון המחקר ב Nationwide Children’s Hospital , הוכיח הישרדות ממושכת והישג במספר רב של אבני דרך שלא נראו קודם לכן במהלך המקובל והטבעי של המחלה SMA1 – מחלה הרסנית, מחלה נוירומוסקולרית מתקדמת, אשר בדרך כלל גורמת למוות התינוק עד גיל 2. הזרקה תוך ורידית של AVXS-101, וירוס המשויך לסרוטייפ (AAV9- Adeno-Associated Virus serotype) העביר את הגן הנוירוני או מה שנקרא SMN- Survival of Motor Neuron הדרוש לטיפול בתינוקות אלה.
הניסוי מבוסס על כמעט 30 שנה של מחקר ושיתוף פעולה יסודי. Arthur Burghes, PhD, מאוניברסיטת אוהיו, יצר את מודל העכברים עם SMA שנותר ומשמש עד היום כתקן שבו כל בודקים תחילה את כל הטיפולים הגנטיים. בריאן קספר, PhD, סגן נשיא בכיר ומנהל מדעי בכיר ב- AveXis, חברה לטיפול בגנים קליניים, המפתחת טיפולים לחולים הסובלים ממחלות גנטיות נוירולוגיות נדירות ומסכנות חיים, במהלך עבודתו ב Nationwide Children's, גילה שוקטור AAV9 יכול לחצות את מחסום הדם העובר למוח BBB – Blood-Brain Barrier כאשר מוזרק לתוך מערכת כלי הדם יכול לספק גנים ישירות לנוירונים המוטוריים. מחקר זה פורסם בכתב העת המדעי Nature Biotechnology בשנת 2009 .
"אף אחד מכל המחקרים האלה לא היה אפשרי ללא התגלית הסמינלית ש- AAV9 חוצה את המחסום Blood-Brain Barrier" מנגנון סינון של נימים כדי לשאת דם אל המוח ורקמת חוט השדרה, לחסימת מעבר של חומרים מסוימים, אומר דר' מנדל, פרופסור לפסיכיאטריה, נוירולוגיה, פתולוגיה ופיזיולוגיה וביולוגיה של התא באוניברסיטת אוהיו. .
וקטורי AAV9 הם כלי מבטיח להעברת גנים בטווח רחב של רקמות – אולי בעיקר במערכת העצבים המרכזית ורקמות השריר. חוקרים ב Nationalwide Hospital בונים על ההצלחה הנוכחית של הטיפול הגנטי ב AAV9 במטרה להציע תקווה לחולים ומשפחות עם מחלות הקשורות במערכת העצבים והשרירים neuromuscular.
"מעבר ל- SMA, אנו עובדים על פתרונות לריפוי גנטי למספר עצום של מחלות נוירו-מוסקולריות ונוירולוגיות – מחלת Charcot–Marie–Tooth disease (CMT), מחלת באטן Batten disease, ומחלת ניוון שרירים", אומר קווין פלאניגן, מנהל המרכז לטיפול גנטי. "וככל שאנו מוצאים את הגורמים הגנטיים עבור יותר ויותר מחלות נדירות, אנחנו הולכים לראות גידול במאמצים למצוא את הטיפול הגנטי המתאים."
המטרה הבאה- משפחות של מחלות עם יותר מגורם גנטי יחיד
ריפוי גנטי עבור מחלה מסוימת אינו תרחיש חד-פעמי. מספר סוגים שונים של מוטציות יכולים להשפיע על גן מסוים או קבוצה של גנים המסדירים תהליך חלבונים או תאים.
תסמונת סן-פליפו Sanfilippo Syndrome
תסמונת סן-פליפו , או Mucopolysaccharidosis MPS סוג III, היא היעד של אחת התוכניות בראשות דר' פלאניגן, שהוא גם מנהל המרכז של המחקר (CORT) בניוון שרירים ופיתוח טיפולים בבית החולים לילדים Nationalwide
מרכז CORT-Center of Research Translation הוקם באמצעות מענק של 7.5 מיליון דולר מהמכון הלאומי לבריאות, של המכון לדלקות פרקים ומחלות שרירים ושלד (NIAMS).
ארבעה גנים שונים עשויים להיות מעורבים, וכתוצאה מכך סוגים MPS IIIA דרך IIID. ווקטורים שפותחו במקור במרכז לטיפול גנטי בחולים עם סוגים MPS IIIA ו- IIIB נמצאים כעת בניסויים ב Nationalwide Children's, בראשות דר' פלאניגן ובמימון Abeona Therapeutics.
"בדומה לניסוי ה-SMA1, מוצרים אלה של טיפול גנטי באמצעות ה- AAV9 המכוונים למערכת העצבים המרכזית (CNS) מועברים דרך הווריד, ונראה כי עד היום הם מציגים בטיחות טובה", אומר דר' פלאניגן.
מחלת ניוון השרירים בגפיים LGMD-Limb Girdle Muscular Dystrophy
"מחלת ניוון השרירים (LGMD) היא דוגמה נהדרת עד כמה מסובך באמת פיתוח טיפולים ספציפיים לגן", אומרת Louise Rodino-Klapac, PhD, החוקרת הראשית במרכז לטיפול גנטי במכון המחקר ב Nationalwide. דר' רודינו-קלאפק היא גם הקצינה המדעית הראשית של Myonexus Therapeutics, חברה לטיפול בגנים קליניים המפתחת טיפולים ראשונים מסוג LGMD מסוג 2D, 2B, 2E, 2L ו- 2C המבוססים על מחקר ב- Nationalwide Children's. דר' רודינו-קלאפק עברה באמצע שנת 2018 להיות המנהלת הראשית למחלקת הטיפול הגנטי בחברת Sarepta Therapeutic אשר מתמחה בטיפול גנטי עם פייפליין רחב בתחום וממשיכה להרחיבו ברכישת חברות טיפול גנטי קטנות ומצרפת אותן לפייפיליין המורחב שלה עם השקעה של מאות מליוני דולרים בהקמת מפעל הגדול בעולם לייצור פלסמיד לטיפול גנטי.
צורות רבות של מחלת ה- LGMD נובעות מבעיה במכלול הקשור לדיסטרופין. כל אחת מ 20 תת הסוגים של ה- LGMD יש שכיחות הנעה בין 1 ל 100,000 ועד 1 ל -200,000 בן-אדם והיא נגרמת על ידי מוטציה ייחודית. לכן, כל סוג משנה צריך גישת הטיפול הגנטי המתאים לו בצורה ייחודית.
במעבדה של רודינו-קלפאק, טיפולים גנטיים עבור שש צורות שונות של LGMD2 נמצאים כעת במחקרים קליניים בשלבים השונים. הטיפול ב- LGMD2E נועד לתקן מוטציה של ביתא-סרקוגליקן, והוא הראשון המאושר לניסוי קליני בבני אדם. הניסוי התחיל לגייס חולים בשנת 2018 וכבר התחילו את הטיפול הגנטי בזרוע הראשונה של הניסוי ב-3 החולים הראשונים.
"התוצאות הפרה-קליניות פוצצו אותנו מרוב הפתעה", אומרת דר' רודינו-קלאפק, שהיא גם פרופסור לרפואת ילדים באוניברסיטה לרפואה של אוניברסיטת אוהיו. "בדרך כלל, אנו שמחים אם רק 50 אחוזים של סיבי השריר המטופל הצליח לבטא את הגן החדש המועבר אליו בטיפול הגנטי. אבל בכל המחקרים הפרה קליניים שלנו, מעל ל 95% מסיבי השריר התגלו כמבטאים את הגן. זה גרם לנו באמת תקווה לגבי מה התרופה תוכל לעשות עבור החולים."
לדברי דר' רודינו-קלפאק, ישנן סיבות רבות לכך ששיעור הביטוי היה גבוה כל כך במחקרים הפרה-קליניים הללו, אך היא משערת כי שימוש ב- AAV משלים (אשר שימש גם בניסוי SMA1) וגודל הגן הקטן יחסית עשוי להיות מהגורמים החשובים.
וקטורים עצמאים scAAV הם וקטורים המכילים רצפים משלימים אשר גורמים לחישול ואיחוד ספונטני כנגד זיהום של התא המארח. טכניקה זו פועלת בצורה הטובה ביותר עבור העברת גנים קטנים, כגון הגן SMN המשמש בניסוי SMA1 ואת הגן betasarcoglycan שהוא המטרה בניסוי ה- LGMD2, מאחר ואורך הגן המקסימלי עבור scAAV הוא 2.4 קילו-בתים (kb).
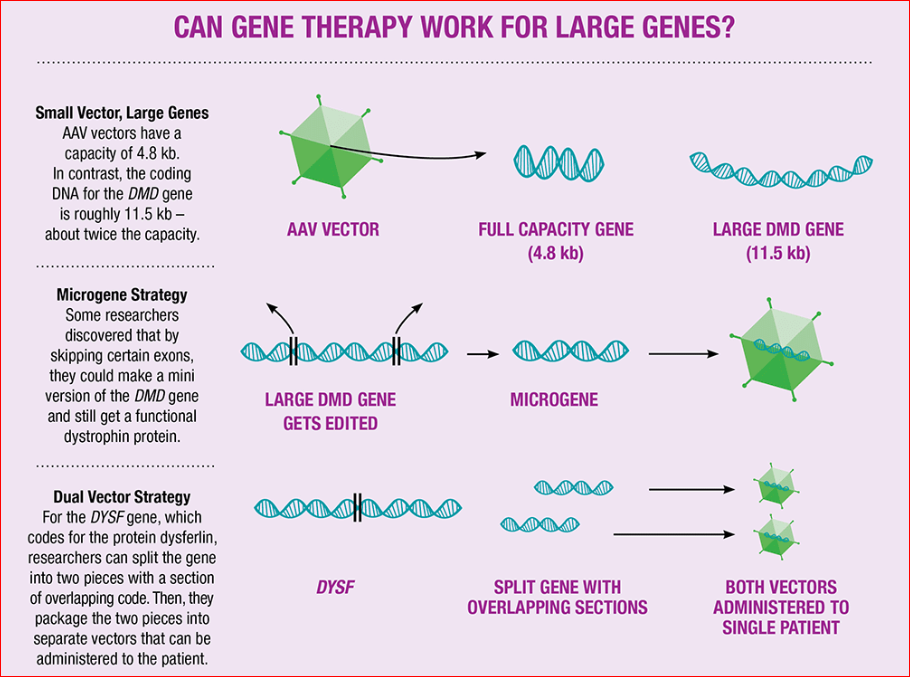
גישות בטיפול הגנטי בעזרת AAV עבור גנים גדולים
אבל מה עם גנים גדולים? בעוד שהטיפול הגנטי בעזרת AAV מוגבל על ידי הקיבולת של הווקטור – רק 4.8kb – קהילת המחקר אינה נרתעת מהתמודדות עם פתרון טיפול גנטי לגן האנושי הגדול ביותר הידוע כיום. טעויות בקידוד הגן עבור דיסטרופין מובילות לניוון שרירי מסוג דושן (DMD), אחד מניוון השריריים הידועים ביותר. גן ה DMD שהוא cDNA הוא בערך 11.5kb – בערך פי 2.5 יותר מאשר הקיבולת המקסימלית של הוקטור.
מיקרו-גנים Microgenes
אמנם, גן ה- DMD גדול מדי כדי להכניס לתוך הוקטור, בשני ניסויים קליניים שנפתחו לאחרונה, החוקרים משתמשים בגרסה מיניאטורית של הגן – מיקרודיסטרופין microdystrophin.
"הרעיון הוא שהגירסה הקטנה יותר של הגן תאפשר לתאים לייצר דיסטרופין 'קרוב מספיק' כדי לשפר את הסימפטומים ואת הישרדותם של הילדים עם ניוון שרירים מסוג דושן – DMD", מסביר דר' מנדל.
ניסוי אחד ב Nationalwide, בראשות דר' רודינו-קלפאק ודר' מנדל, יבדקו את היעילות של מוצר הטיפול הגנטי microdystrophin של חברת Sarepta Therapeutics. ניסוי נוסף, בראשותו של דר' בארי ביירן, מנהל המרכז על שם פאוול לטיפול גנטי באוניברסיטת פלורידה, בוחן גרסה שונה במקצת של microdystrophin המיוצר על ידי Solid Biosciences.
"בעבודתנו הפרה-קלינית, המיקרודיסטרופין היה יעיל בהפקת פנוטיפ קרוב לטבעי", אומרת דר' רודינו-קלאפק. "אנו מקווים להשפעה עמוקה של הניסויים הקליניים בשלב 1, אך נצטרך לראות עד כמה יעיל המיני-גן בהשוואה לזה שבאורך מלא".
"עד לנקודה זו, היה לנו מעט מאוד להציע עבור המשפחות האלה, "אומר דר' מנדל. "במשך 50 שנים, הטיפול המאושר היחיד שלנו עבור דושן -ניוון שרירים היה Prednisone. מטרת המחקר שלנו היא לבדוק האם טיפול גנטי הוא אפשרות בטוחה ויעילה עבור חולים אלו ואחרים עם מחלות נוירומוסקולריות נדירות בעתיד ".
הניסוי הקליני של Nationwide עם ה- microdystrophin עבור DMD יהיה השימוש הראשון של טיפול גנטי תוך ורידי לכל סוגי ניוון השרירים, על פי דר' רודינו-קלאפק. "נתחיל עם ילדים בגילאי 4-7 שנים. ילדים אלה עדיין בשלב מוקדם בתהליך המחלה, ואנו מקווים כי על ידי התערבות מוקדמת, אנחנו יכולים לעצור את התקדמות המחלה. קבוצה שנייה, עם ילדים בגילאי 3 חודשים עד 3 שנים, תאפשר לנו לראות אם נוכל למנוע את הופעת הסימפטומים באמצעות הטיפול הגנטי ".
הצוות ינהל מינונים שהיו יעילים במחקרים פרה-קליניים. קבוצת ביקורת של חולי DMD תקבל פלסבו ויבוצע מעקב אחרי החולים במשך שנה אחת. אותם חולים אשר היו בזרוע הפלסיבו יקבלו טיפול גנטי בשנה השנייה של הניסוי.
הניסוי הנוסף של אוניברסיטת פלורידה, המגייס כעת משתתפים, פתוח לילדים בגילאי 4 עד 17 שנים ומשתמש בקבוצת ביקורת. "המחקר שלנו עבור ניסוי זה מאפשר לנו להתאים בקרות בנוסף לקבוצת הטיפול", אומר דר' ביירן, גם פרופסור לרפואת ילדים וגנטיקה מולקולרית ומיקרוביולוגיה באוניברסיטת פלורידה. לחולי הביקורת תהיה הזדמנות לקבל את הטיפול מאוחר יותר, בהתבסס על עיצוב התחלה מאוחרת. זה מספק השוואה סבירה של תופעות לוואי ויתרונות של הטיפול. "
האם שני וקטורים יכולים להיות טובים יותר מאחד?
בדומה לגן ה- DMD, הגן לדיספרלין (DYSF) גדול מדי מכדי להכניס בוקטור AAV. עם זאת, באמצעות טכנולוגיית וקטור כפול כדי להגדיל את קיבולת האריזה של ה- AAV, החוקרים פיתחו חפיפות dysferlin. טכניקה זו מסתמכת על רקומבינציה הומולוגית כדי להרכיב שני פלסמידים המועברים בוקטורים נפרדים בחזרה יחד עם ההגעה לתא המטרה.
"גן המטרה, במקרה זה, DYSF, מחולק בין שתי פלסמידים להעברה עם רצף משמעותי בחפיפה", אומרת דר' רודינו-קלאפק. "כאשר מוזרקים יחד, שני הוקטורים נכנסים לתא והפלסמידים מתחברים יחד בכדי לבטא את הגן באורך מלא.
"עבודה פרה קלינית רחבת היקף עם טכנולוגיה זו עבור גנים אחרים מראה תוצאות מעורבות, כאשר החשש העיקרי הוא היעילות הנמוכה של הרקומבינציה או חיבור שני חלקי הגן. עם זאת, החוקרים מקווים שזה יהיה פתרון יעיל עבור כמה סוגי גנים. במקרה של DYSF, זה עבד טוב להפליא, אומרת דר' רודינו-קלאפק.
דר' רודינו-קלפאק מובילה מחקר תוך שרירי תוך שימוש בגישה וקטורית כפולה לדיספללינופתיה dysferlinopathy – דיסטרופיה שרירית המשפיעה על מטופלים בשנות העשרה המאוחרות או בתחילת שנות ה -20, כאשר כשליש מהחולים נדרשים לכסא גלגלים בתוך 15 שנים מהאבחנה. LGMD2B ו Miyoshi myopathy הן שתי צורות הנפוצות ביותר של dysferlinopathy.היא מקווה כי ניסוי קליני תוך ורידי יכול להתחיל בשנת 2019.
לעמוד באתגרי עתיד הטיפול הגנטי בעזרת AAV
"בימים אלה, שיקול אחד קריטי בניסויים קליניים אלה הוא, המינון הנוכחי, שהוא מינון חד-פעמי עבור הילדים האלה. נכון לעכשיו, לא תהיה להם הזדמנות למינון נוסף חזק יותר. ברגע שהם מוזרקים עם הוקטור, הגוף מתחיל לייצר נוגדנים וחסינות אליו ומינונים עתידיים לא יהיו יעילים ", אומר דר' מנדל.
כמו כן, אם למישהו יש חשיפה סביבתית ל- AAV9, ייתכן שיש להם נוגדנים שימנעו מהם להשתתף בניסוי הקליני, בשל הסבירות שהטיפול לא יוכל לעבור את המערכת החיסונית.
"שובר את הלב להוציא חולים מניסוי קליני כי יש להם נוגדנים ל- AAV", אומרת דר' רודינו-קלאפק. "התקווה שלנו היא שתהיה לנו אסטרטגיה להציע את הטיפול בבטחה וביעילות לחולים עם חשיפה סביבתית לוירוס".
דר' מנדל ורודינו-קלפאק ועמיתיהם במרכז לטיפול גנטי ב- Nationalwide Children's הצליחו בניסיון בשימוש בתרופות אפרהזיס apheresis ותרופות אימונוטרפוטיות במודלים של בעלי חיים. השלב הבא יהיה לעצב ניסוי קליני לבדיקת התהליך בחולים.
"פתרון הבעיה של מינון-חוזר הוא חיוני כדי להציע את התוצאות הטובות ביותר עבור חולים ובני משפחותיהם", אומר דר' מנדל. "פיתוח הפתרון הוא הדבר הנכון לעשות".
צעד נוסף בדרך לעתיד של הטיפול הגנטי הוא בדיקות תינוקות שרק נולדו. בדיקות ילוד מגדירות מחלות גנטיות רבות שהטיפול הגנטי שואף לטפל בהן. וככל שיותר טיפולים גנטיים יהיו זמינים, מספר המחלות הגנטיות ברשימת הבדיקה המוקדמת צפוי לגדול. אז מתי צריך לתת טיפול גנטי?
"אנחנו מאמינים שככל שהילד יוכל לקבל טיפול מוקדם יותר, התוצאה תהיה טובה יותר", אומר דר' מנדל. "בניסוי שלנו, החולים עם SMA1 שטופלו בשלב מוקדם מגיעים לאבני דרך שלא נראו בהיסטוריה הטבעית של המחלה".
כאשר לטיפול הגנטי יש את היכולת לא רק לעצור את התקדמות המחלה, אלא אם ניתנה מוקדם מספיק – לעצור את תחילת הסימפטומים לחלוטין, נראה שלשאלה יש תשובה ברורה. עם זאת, לאור העדר מחקרים ארוכי טווח עבור טיפולים חדשים אלה, החוקרים עדיין לא יודעים כמה זמן תימשך ההשפעה של הטיפול.
"זה חלק מהשיחה על המינון מחדש", מוסיף דר' ביירן. "כמו מחלות עם השפעה בשלב מאוחר בחיי החולה מזוהים בתחילת החיים, אף אחד לא בטוח מה לעשות עם המידע הזה. אבל נראה כי התערבות מוקדמת תוביל לתוצאות טובות יותר ".
טיפול בילדים קטנים יותר דורש גם פחות וקטור. במחקר SMA1, החוקרים נתנו את הטיפול הגנטי לתינוקות. בניסויים הבאים של DMD ו- LMGD, המשתתפים יהיו בטווח של 4 עד 12 שנים.
"הדרישות של שימוש בטיפול גנטי עם AAV בחולי DMD המבוגרים יותר, וגוף גדול יותר, מותחים את המשאבים של הטכנולוגיה המתקדמת", אומר דר' ביירן.
בעוד החוקרים הראו בטיחות ויעילות של AAV9 במחקרים פרה קליניים רבים, ובמספר קטן של ניסויים קליניים בשלב מוקדם, גבולות הטכנולוגיה טרם הוגדרו. במחקר שפורסם לאחרונה ב Human Gene Therapy, ג'יימס ווילסון, PhD, ועמיתיו מדווחים על רעילות חמורה במודלים של בעלי חיים גדולים, בעקבות מינון גבוה של IV הזרקה לוריד במינון גבוה של וקטור AAVhu68 המבטא את ה- SMN האנושי. במהלך הניסוי של SMA1 של Nationwide לילדים, ארבעה חולים סבלו מאנזימי כבד מוגדלים אסימפטומטיים שהצריכו טיפול ב prednisone. החוקרים מציינים כי לא נצפו תופעות לוואי נוספות במהלך הניסוי.
שני גורמים עשויים להסביר את ההבדל הבולט בתוצאות שני המחקרים. ראשית, וקטור המשמש במחקר של דר' וילסון הוא מוטציה AAV9, ובעוד זה רק שני זוגות בסיס שונה מסרוטיפ AAV9, זה לא עבר את אותה בדיקה כמו AAV9, אומר Jaysson Eicholtz, מנהל GMP (ייצור בתהלכים מאושרים) ב Nationwide Children’s. בנוסף, שיטות שונות שימשו לכימות ריכוזים של וקטור המשמש בטיפול גנטי המיוצר בכל מתקן ייצור קליני (CMF).
"אין לנו תקן בתעשייה עבור תהליך זה, אבל ב Nationalwide אנו משתמשים בתהליך ידני אשר היה עקבי בכל המחקרים, הפרה קליני, רעילות ויצור מוצרים קליניים סופיים", אומר Eicholtz. "שיטת ה- PCR הדיגיטלית המשמשת במוצרים שבדו"ח של דר' ווילסון עלולה לגרום לריכוזים של וקטור פי ארבעה מהשיטה בה משתמשים ב- Nationwide Children עבור המינון".
דר' ביירן מציע כי פיתוח דרכים טובות ועקביות יותר למדוד ריכוזים ברחבי התעשייה יהיה צעד חשוב בהבאת טיפול גנטי לניסויים קליניים. "במובן התיאורטי, אתה יכול לעשות את החישוב להגדלת הייצור… אבל עד שאתה באמת עושה את זה, יש הרבה שאתה לא יודע", הוא אומר. "יש לנו הרבה עדיין עבודה רבה בפיתוח אנליטיקה עבור bioavailability ועבור הפוטנציה. שאלות אלו סווגו בעולם המולקולה הקטנה, אך עדיין אין לנו עקביות לבדיקות אלו בעולם הטיפול הגנטי".
אם המדע מוכן, האם השוק יבין את הפוטנציאל ויגלם אותו?
באקלים הפוליטי והכלכלי של היום, כל התקדמות רפואית מתקבלת תחילה בהתרגשות ובאופטימיות, ומיד אחר כך עולה השאלה "כמה זה יעלה?" חודש לאחר האישור, חשפה חברת ספארק תרפויטיס Spark Therapeutics שהיא תגבה 850 אלף דולר עבור מינון חד-פעמי של טיפול גנטי באובדן הראייה לכל עיין (או 1.7 מליון דולר לשתי העיניים). זה תג מחיר גבוה אשר הופך אותה לתרופה היקרה ביותר שנמכרה בארצות הברית. כמובן, המשא ומתן עם חברות הביטוח לוקח את הדיון על התמחור קדימה, כולל החזרים מבוססי תוצאות.
"אלה טיפולים יקרים להפליא לייצר", אומר דר' פלאניגן, שהוא גם פרופסור לרפואת ילדים ונוירולוגיה באוניברסיטת אוהיו סטייט קולג 'לרפואה. "אנחנו נצטרך להבין אם המודל שלנו עובד גם בטווח הארוך. ככל שאנו צוברים יותר ויותר מחלות נדירות הניתנות לטיפול על ידי טיפול גנטי, עלויות מעל המיליון דולר למינון לא יכולות להיות בנות קיימא ".
תרומה ישירה לתמחור היא האתגר למסחר ולייצור המוני של התרופות לטיפול גנטי. הגדלה הדרגתית וייצור הן נקודות מפתח בשיחות עם שותפים בתעשייה, אומרת דר' רודינו-קלאפק. "זה קצת שונה לייצר מספיק וקטור עבור מאות חולים לעומת תריסר חולים בניסוי קליני.
"כאשרו מוצרים מבוססי וקטור יתחילו להיכנס לשוק, שותפים בתעשייה יידרשו להתמודד עם האתגרים של קנה המידה וההפצה.
"אני אופטימית שנפגוש את האתגרים שהיו לנו. כשהגעתי לראשונה ל- Nationwide Children's, היינו צריכים להבין איך אנחנו יכולים לעשות מספיק וקטור שיספיק בכדי לעשות ניסוי קליני IV. ובכן, עכשיו אנחנו כאן, ואנחנו עושים את זה בימים אלה. זה לא בלתי נסבל ", אומרת דרק' רודינו-קלאפק. "אם המדע נמצא שם, והתרופה יעילה, נמצא דרך … איך לא?"
Image credits: Nationwide Children’s